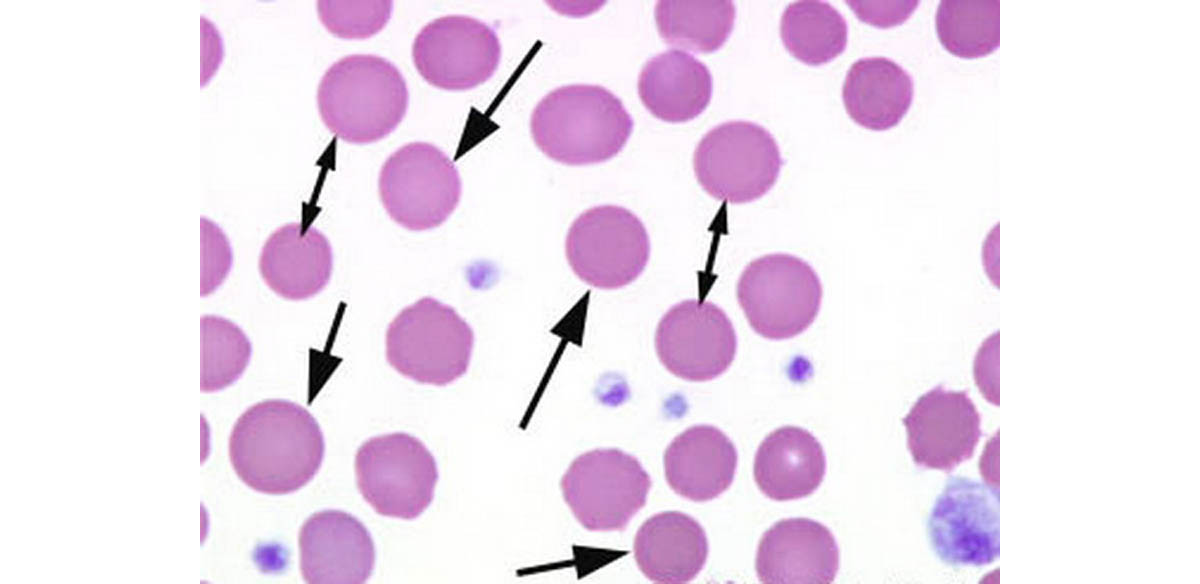
The most common characteristic of hereditary spherocytosis is a structure called microspherocyte – the sphere-shaped red blood cell, caused by a loss of membrane surface area, and an abnormal osmotic fragility in vitro. Because spherocytes have a smaller surface area through which oxygen and carbon dioxide can be exchanged, they perform adequately to maintain healthy oxygen supplies. However, they have a high osmotic fragility – when placed in water, they are likely to burst.
Frequency of the condition
Hereditary spherocytosis is the most common of all hereditary hemolytic forms of anemia. In the United States, the incidence of the disorder is approximately 1 case in 5,000 people. This condition is usually transmitted as an autosomal dominant trait, and the identification of the disorder in multiple generations of affected families is the rule. An autosomal recessive mode of inheritance also occurs. The disease is also encountered worldwide, but its incidence and prevalence in other groups are not established clearly. Anemia or hyperbilirubinemia may be of such magnitude as to require exchange transfusion in the neonatal period. Anemia is usually mild to moderate; however, sometimes it is very severe and sometimes not present at all. Splenomegaly is the rule, and palpable spleens have been detected in more than 75% of affected subjects. Severe hemolytic anemia requires red cell transfusions.
Mechanism of the condition
The defects in hereditary spherocytosis are in the red cell membrane; proteins essential for the integrity of the membrane structure lie immediately under the lipid layer.
Different genes code each of these proteins, thus hereditary spherocytosisis a heterogeneous disorder, which can result from a defect in any one of these proteins.
Symptoms of the condition
The spleen's hemolysis results directly in varying degrees of anemia and hyperbilirubinemia.
These two conditions can result in symptoms of:
- Fatigue
- Pallor
- Jaundice
Chronic symptoms include:
- anemia
- splenomegaly (enlargement of the spleen)
Also, the detritus of the broken blood cells accumulates in the gallbladder, and can cause gallstones to develop.
Differential diagnosis
This condition can often be misdiagnosed with several other conditions and the most common ones are:
- Anemia
- Biliary Colic
- Biliary Disease
- Biliary Obstruction
- Cholecystitis
- Cholelithiasis
- Hemolytic Anemia
- Hyperbilirubinemia, Conjugated
- Hyperbilirubinemia, Unconjugated
Diagnosis
There are several possible methods and diagnostic tests available to get to the proper diagnosis.
Some of the most commonly used are:
Lab Studies: The classic laboratory features of this type of anemia include minimal or no anemia, reticulocytosis, an increased mean corpuscular hemoglobin concentration (MCHC), spherocytes on the peripheral blood smear, hyperbilirubinemia, and abnormal results on the osmotic fragility test.
Other biochemical changes of hemolysis also are also present, including:
- increased lactate dehydrogenase (LDH)
- increased unconjugated bilirubin
- decreased serum haptoglobin
- an increase in MCHC
Other laboratory tests used to diagnose this type of anemia include the autohemolysis test and the glycerol-lysis test. The initial workup if hemolysis is suggested should include the following:
- Reticulocyte count
- Lactate dehydrogenase
- Fractionated bilirubin
- Haptoglobin
- Peripheral smear with megalocytosis
- Vitamin B-12 and folate
- Herpes simplex virus, HPV type 19, and infectious mononucleosis
Hb
Dec (Reflects EVH)
RCC
Dec (Reflects EVH)
Hct
Dec (Reflects EVH)
WCC
N - Inc
Plt
N - Inc
MCV
N - Inc (Microspherocytes have normal volume)
MCH
N - Inc
Haemoglobinisation
Normochromic
Anisocytosis
++ (Polychromatic Macrocytes)
Poikilocytosis
++ - +++ (Microspherocytes)
Immature Forms
Polychromasia + - +++ (Nucleated RBCs if severe)
Imaging Studies: If the patient displays signs and symptoms of hemolysis in addition to right upper abdominal quadrant pain, an ultrasound of the biliary tree should be performed to help exclude cholecystitis or cholelithiasis.
Procedures: If an aplastic crisis is suggested, further evaluation and platelets should be done. This may require a bone marrow biopsy and aspirate to rule out aplasia or megaloblastosis. Obtaining bone marrow aspirate for testing is rarely necessary except in cases of aplastic or megaloblastic crisis.
Histological Findings: Anemia, reticulocytosis, and spherocytosis on peripheral blood smear examination provide strong proof required to set an accurate diagnosis of this type of anemia. Autoimmune hemolytic anemia also may produce spherocytosis, which usually can be excluded by negative findings on a direct anti-globulin test.
Medical Care – Splenectomy
The treatment of this type of anemia involves pre-splenectomy care, splenectomy, and optionally, post-splenectomy complications. Children with severe hyperbilirubinemia caused by this type of anemia are at risk for kernicterus; these infants should be treated with phototherapy and exchange transfusion as clinically indicated.
Splenectomy indications
Indications for splenectomy are not always clear. Doubt exists because the patients with more severe anemia and symptoms and complications of this spherocytosis could have many complications after the splenectomy. Similarly, splenectomy can be deferred safely in patients with mild uncomplicated HS. Also, no good studies have been performed that provide a basis for clinical judgments in patients with this hereditary spherocytosis
Splenectomy is usually curative
Red cell survival improves significantly, but not to normal levels. Post-splenectomy blood changes include an increased hemoglobin level, decreased reticulocyte count, and the appearance of specific inclusion bodies and target cells.
There is a possibility of developing fatal sepsis caused by capsulated organisms. This complication is recognized in children who have had splenectomy performed.
A simultaneous cholecystectomy in patients with bilirubin stones may eliminate future complications.
Bilirubin gallstones
Bilirubin gallstones are found in approximately 50% of patients with hereditary spherocytosis. They are frequently present in patients with very mild forms of the disease. Performing a prophylactic laparoscopic cholecystectomy seems reasonable in such cases. This procedure helps prevent significant biliary tract disease and, in some patients with mild HS, helps avoid the need for splenectomy.
Splenectomy for children with hereditary spherocytosis should be performed only if the child is over 6 years old.
Oral folate supplements
Researches have shown that most children in developed countries consume well above the minimum daily requirement of oral folate supplements. However, there is no strong evidence to support universal folate supplementation in hereditary spherocytosis, and it is likely that it is only required in children with severe and moderate HS, and for all patients during pregnancy.
Iron supplementation
Iron supplementation supports the increased production of red blood cells, but in longstanding cases in which patients have taken supplemental iron or received numerous blood transfusions, iron overload may be a significant problem, being a potential cause of cardiomyopathy and liver disease.
Conclusion
Hereditary spherocytosis is the most common form of haemolytic anaemia seen in northern Europe. Most children which have a mild form of the disease can live a normal life, and do not require a splenectomy. Splenectomy is reserved for those with severe cases, or those who develop symptomatic gallstones.
- Photo courtesy of isis325 by Flickr : www.flickr.com/photos/92708411@N07/8550844204/
- www.adc.bmjjournals.com
- en.wikipedia.org/wiki/Hereditary_spherocytosis
- image: hereditaryspherocytosis.org