Table of Contents
One out of every 500 African-American births. One out of every 36,000 Hispanic-American births. Approximately 90 to 100 thousand Americans suffer from the pain, genetic disorder known as sickle cell disease. The rate of those carrying the sickle cell disease trait is even higher, with 1 in 12 African-Americans inheriting the sickle cell gene from one parent. Yet despite these high rates, people with sickle cell disease are less likely to have access to comprehensive care than those with other genetic disorders, such as cystic fibrosis and hemophilia. What is sickle cell disease? How can those diagnosed obtain the best care possible?
An Inherited Disorder
Sickle cell disease is an autosomal recessive trait that affects the hemoglobin in red blood cells. An autosomal recessive disorder means that two copies of the gene, one from each parent, must be passed down for the disease to develop in a child. If only one gene is passed to the child, he would be a carrier, but would have no symptoms. With one out of 12 African Americans as carriers, it is easy to see why this community in particular is hardest hit by sickle cell disease.
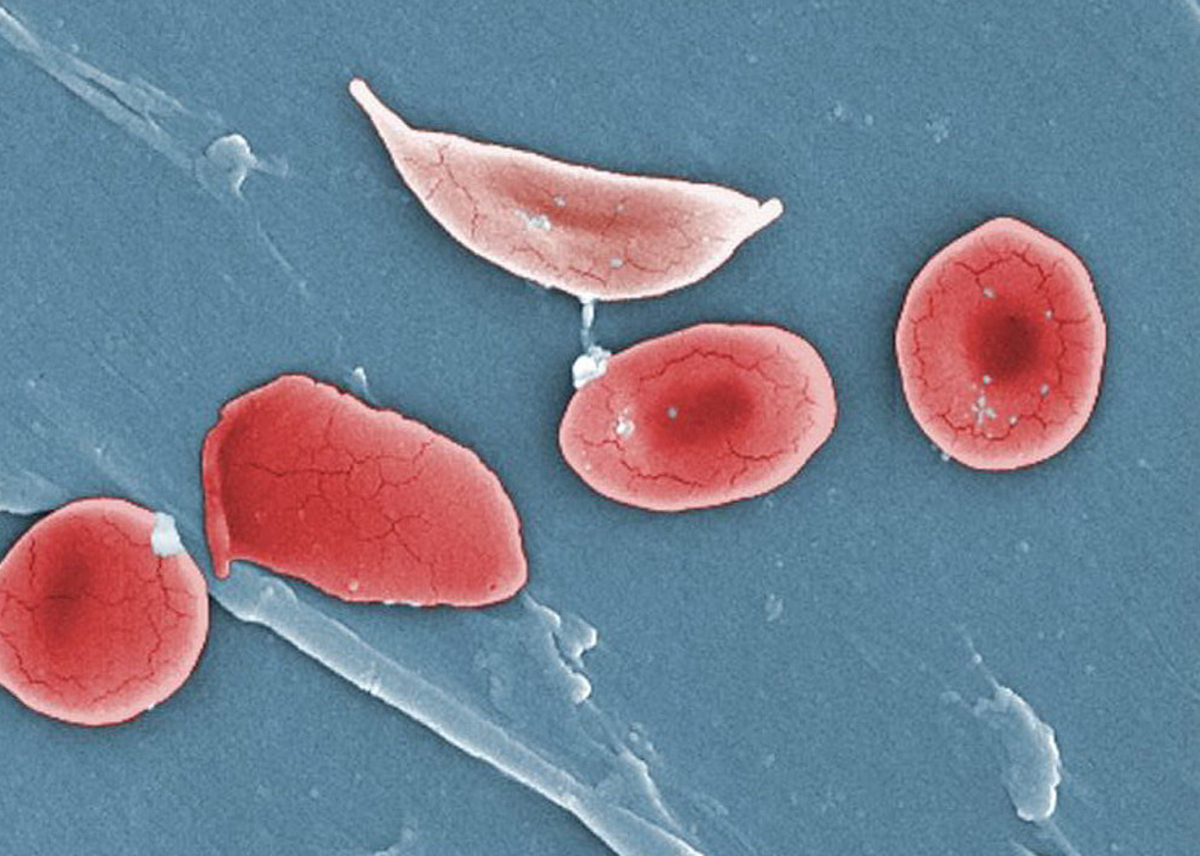
Sickled Cells
The disease gets its name from the appearance of the red blood cells of those affected. Instead of the usual doughnut-like shape, the red blood cells of those with sickle cell disease become hard, sticky, and c-shaped. These changes allow the cells to clump together and impede blood flow throughout the body. The deformed cells also die more quickly. Normal red blood cells have an average lifespan of 120 days and are constantly replenished by the bone marrow.
The Symptoms of Sickle Cell Disease
The chronic anemia caused by the shortened lifespan of sickled cells means that the person affected is constantly low on oxygen. Normal red blood cells combine oxygen with hemoglobin to transport around the body. With abnormal hemoglobin, those with sickle cell disease display multiple symptoms of hypoxia, or oxygen deficiency, such as pale skin, weakness, and fatigue. The high levels of red blood cell destruction also can result in jaundice as the sickled cells fragment and release bilirubin into the blood stream. Jaundice is evident in the yellowing of the skin, although in those with darker complexions it is more easily seen as yellowing of the sclera, or white part of the eye.
It is the shape of the cells themselves, however, that can lead to the most severe symptoms of sickle cell disease. Known as a sickle cell crisis, the clumping of the abnormal red blood cells and their fragmentation results in occlusion of the veins and arteries. This blocking of the small blood vessels can cause sudden, intense pain that can last anywhere from hours to days or even longer. While sickle cell disease is usually managed by a person's family practice doctor, the crises are the top reason why people seek help from a hospital emergency room.
Read More: National Aplastic Anemia and MDS Awareness Week
Getting Through a Crisis
When a person develops a sickle cell crisis, the symptoms are severe. The normal pallor of a person with sickle cell disease becomes even more pronounced, with pale lips, tongue, palms of the hands, and nail beds. It's difficult to wake him and, even when awake, he is listless and lethargic. He is irritable, understandable considering that a crisis involves severe pain. A high temperature, one over 104 degrees Fahrenheit, is present for at least two days and sometimes more.
- Anemia, sickle cell. (2001). In Taber's Cyclopedic Medical Dictionary (pp.105-106, Edition 19). Philadelphia, PA: F. A. Davis Company.
- Photo courtesy of Anatomy & Physiology, Connexions Web site by Wikimedia Commons : en.wikipedia.org/wiki/File:1911_Sickle_Cells.jpg
- Photo courtesy of Nephron by Wikimedia Commons : commons.wikimedia.org/wiki/File:Sickle_cell_disease_and_cirrhosis_-_high_mag.jpg
Your thoughts on this