When a person is diagnosed with amyotrophic lateral sclerosis, the world is changing. They are suddenly faced with a serious disease that could affect every aspect of their life. Therefore, it is clear these patients need help, especially those who diagnosed with ASL later in life. Answering some frequently asked questions they might have would be a good start.
What is amyotrophic lateral sclerosis?
The word “amyotrophic” means “without muscle nourishment”. It refers to the loss of signals the nerves normally send to the muscles.
“Lateral” means “to the side”, and refers to the location of the damage in the spinal cord.
“Sclerosis” means “hardened” and refers to the hardened nature of the spinal cord in advanced ALS.
ALS attacks the nerve cells, or neurons, responsible for controlling voluntary muscles. In ALS, both the upper motor neurons and the lower motor neurons degenerate or die. Off course, once this happen, neurons cease to send messages to muscles. Unable to function, the muscles gradually weaken, waste away, and twitch. Eventually, the ability of the brain to start and control voluntary movement is lost as well. Individuals with ALS lose their strength and the ability to move their arms, legs, and body, which can be frightening. When muscles in the diaphragm and chest wall fail, individuals lose the ability to breathe without artificial support.
In most cases, the disease does not impair a person’s mind, personality, intelligence, or memory. It will probably not affect a person’s ability to see, smell, taste, hear, or recognize touch. A small percentage of patients may experience problems with memory or decision-making. There is growing evidence that some may even develop a form of dementia caused by ALS.
Who gets ALS?
ALS usually strikes in late middle age or later in a person’s life, although there have been cases of ALS in young adults and even in children, as well as in the very elderly. Some genetic forms of ALS have their onset in youth as well. Men are slightly more likely to develop ALS than women are. Studies suggest an overall ratio of about 1.2 men to every woman.
Genetic factors are part of the picture in ALS, and the disease can run in families. For years, experts have tried to find the factors common to people who develop ALS. They were searching some environmental toxins, occupational hazards, places of work or residence, and so forth. So far, the evidence of such risk factors and triggers has been frustratingly unclear, although a recent finding of an association between developing ALS has indicated one of the strongest of these proposed risk factors.
Does ALS run in families?
ALS does indeed run in families, showing a family history about 10 percent of the time. Several genes associated with ALS are identified, or at least mapped to a specific region of a chromosome.
What happens to someone diagnosed with ALS?
In ALS, nerve cells that control the muscles are gradually lost. In most cases, the cause remains unknown. As these motor neurons are lost, the muscles they control weaken. As a logical consequence, they became nonfunctional after a while. Eventually, the person with ALS might be paralyzed. Death, usually from respiratory complications, typically comes between three and five years after the diagnosis, although the timing is not entirely clear. About 10% of people suffering from this disease live more than 10 years, and some survive for decades. Modern technology has allowed people with ALS to compensate for almost every loss of function to some degree, making it possible even for those with almost no muscle function to continue to breathe, communicate, move about and use a computer. Longevity statistics may be somewhat out of date because of the changes in supportive care and technology developed lately. It is important to note that ALS does not directly affect the involuntary muscles. The most important involuntary muscles are those that control the heartbeat, gastrointestinal tract. and bowel function, bladder and sexual functions. However, prolonged inability to move and other effects of ALS can have some indirect impact.
Pain is not a major component of the disorder, although moderate pain can certainly occur as a result of immobility and its various complications. Hearing, vision, touch and intellectual ability generally remain quite normal in patients diagnosed with ALS. Some experts believe that certain emotional changes may attributes directly to the disease process. However, in such devastating disorder as ALS, it can be difficult to distinguish feelings due to the underlying disorder from those that result from the person’s situation.
What does ALS do to the nervous system?
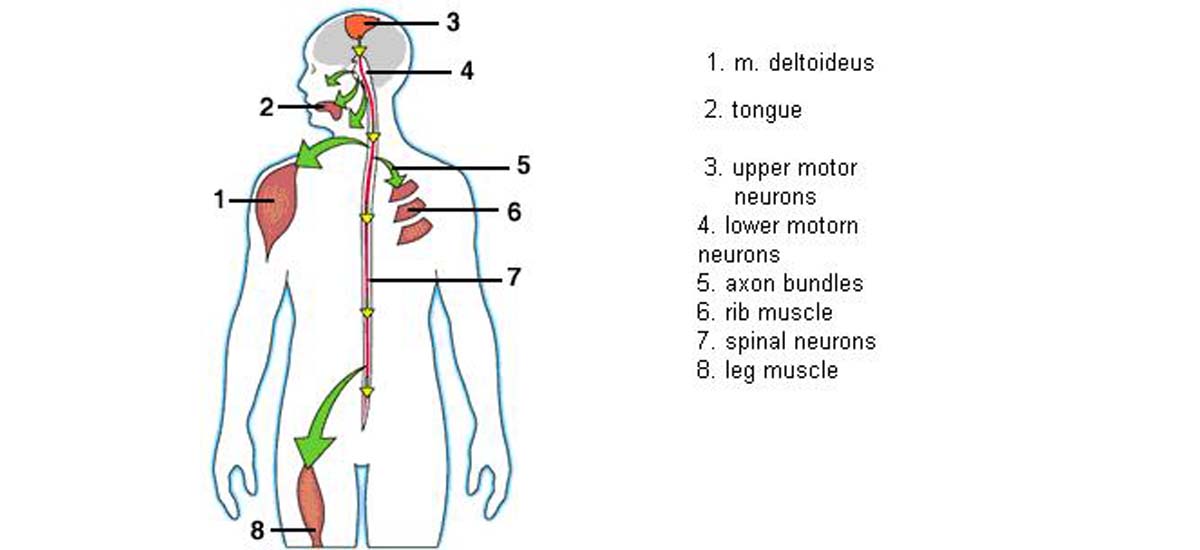
We can divide the muscle-controlling nerve cells, or motor neurons, into two types. The upper motor neurons are located in the upper part of the brain and exert some control over the lower motor neurons in the brainstem and the spinal cord. The lower motor neurons are attached to muscles through wires called axons. Bundles of these axons leave the spinal cord and extend out to the muscles all over the body. (These are the bundles that doctors are referring to when they talk about the nerves.) The function of lower motor neurons is straightforward; they send “go” signals to muscles. When these cells gradually die, as is the case with ALS, muscles become progressively weaker and eventually unable to move. This causes paralysis in patients.
The lower motor neurons that control most of the body are in the spinal cord, and those that control the muscles of speaking, swallowing, and facial expression are in the brainstem. Another name for these is bulbar motor neurons, because the part of the brainstem that houses them has a bulblike shape. The term “bulbar involvement” means that the muscles of the face, mouth and throat are involved in the ALS disease.
The upper motor neurons have more complex functions. It is harder to study them, and there is much, we don’t understand about them, although some new techniques are gradually changing that. These cells seem to exert complex control over the lower motor neurons that allow movements to be smooth. When upper motor neurons are lost and lower motor neurons remain, movements are still possible. However, these movements can become spastic. Beside this symptom, affected muscles became less precise. In ALS, a combination of these effects is usually present, because both upper and lower motor neurons are dying. People with ALS can have weak and wasted muscles combined with tightness. Muscle twitches and cramps are common, and they occur because degenerating axons of the nerves become irritable.
Is there any treatment for ALS patients?
There is no cure for ALS at this moment. However, the FDA has approved the first drug treatment for the disease, and that is Riluzole. Riluzole reduces damage to motor neurons and can prolong survival by several months, mainly in those with difficulty swallowing. Other treatments focus on relieving symptoms and improving the quality of life for people with ALS. Drugs are also available to help individuals with pain, depression, sleep disturbances, and constipation as the main ALS symptoms. Individuals with ALS may eventually consider forms of mechanical ventilation or respirators.
What is the prognosis for those newly diagnosed with ALS?
Regardless of the part of the body first affected by the disease, muscle weakness and atrophy eventually spread to other parts of the body as the disease progresses. Individuals with ALS face increasing problems with moving, swallowing, and speaking or forming words. Eventually, people with ALS will not be able to stand or walk, get in or out of bed on their own, or use their hands and arms, and in the later stages of the disease, they will experience breathing difficulties as the muscles of the respiratory system weaken. Although ventilation support can ease problems with breathing and prolong survival, it does not affect the progression of this disease. Unfortunately, most people with ALS die from respiratory failure, usually within 3 to 5 years from the onset of symptoms. However, about 10 percent of those afflicted with ALS survive for 10 or more years, so death is not the only option.
New research
The National Institute of Neurological Disorders and Stroke conducts research in its laboratories at the National Institutes of Health. They also support additional research through grants to major medical institutions across the country. The goals of this research are to find the cause or causes of ALS. The goal is also to understand the mechanisms involved in the progression of the disease, and develop effective treatments. A key factor may be the Muscular Dystrophy Association, which offers the best doctors and health care professionals.