Sickle cell anemia is a genetic blood disorder wherein the person affected produces abnormal red blood cells containing hemoglobin S instead of hemoglobin A. When the red blood cells with hemoglobin S lose their oxygen they become distorted and adopt a crescent/sickle shape. These cells are usually rigid and sticky and may get stuck in small blood vessels, thus slowing or blocking blood flow as well as oxygen delivery to many parts of the body.
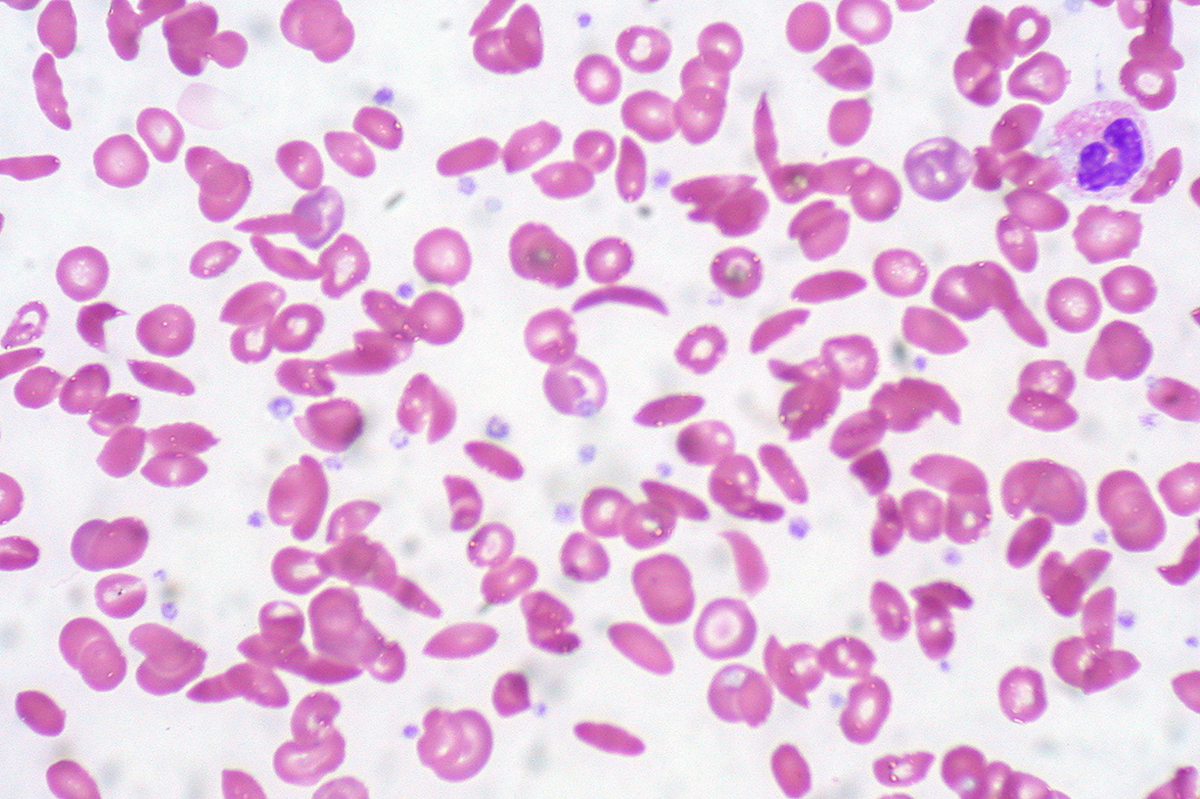
Sickle Cell Anemia is also known as hemoglobin SS disease (Hb SS) or sickle cell disease. It is usually common in people of Mediterranean or African descent.
Symptoms Of Sickle Cell Anemia
Symptoms of Sickle Cell Anemia usually do not occur until an infant is four months old. Common symptoms may include:
- Anemia: The sickle cells are rigid and fragile and can break easily. Normal red blood cells usually die after 120 days of being produced but the sickle shaped cells die in less than 20 days. This results in the shortage of red blood cells (anemia) and cause chronic fatigue and exhaustion.
- Crises: Crises are termed as the episodes of pain and are a major symptom of sickle cell disease. This pain develops as a result of sickle shaped red blood cells blocking the small blood vessels that reach to the chest, abdomen, joints, lower back and even bones etc. Duration and intensity of pain may vary from person to person. Some people may have one episode of pain in a year and some may have many which may require hospitalization. This crisis may be:
- Swollen hands, feet or abdomen
- Vision Problems ranging from poor eyesight to blindness
- Ulcers
- Small strokes which may result in problem in thinking, numbness, trouble walking or talking, weakness in face, arms or legs
- Delayed growth or puberty
- Joint pains
- Infection (gall bladder, urinary, lung, bone etc) resulting in fever.
Sickle Cell Anemia: Diagnosis
Screening test
A blood test can easily determine if a person is affected by sickle cell disease or the sickle cell trait with the presence of hemoglobin S in red blood cells. An affected newborn is usually put under daily antibiotics treatment to avoid any infection. Prenatal diagnosis can also be done by testing the amniotic fluid for the presence of the sickle cell gene.
See Also: Hope for a Cure: Sickle Cell Disease
High risk of stroke is a serious threat to the life of patients with sickle cell anemia. The following tools are available for diagnosing the risk of a stroke:
- TCD (Transcranial Doppler) ultrasonography: The ultrasonography measures the blood flow in the brain and may identify children who are at risk of a stroke. But even if the screening diagnosis shows a normal blood flow velocity, high risk children may still be at risk of a stroke.
- MRI (Magnetic Resonance Imaging): It detects small blockages in blood vessels.
- Angiography: A technique for detecting aneurysms (a bulging in blood vessel wall, which if it bursts in the brain may result in a stroke).
- Genetic Markers: Researchers are also looking for possible genetic markers which may identify people with sickle cell disease at higher risk of strokes.
Treatment Of Sickle Cell Anemia
Existing treatments of sickle cell anemia are usually aimed at avoiding crises and relieving the symptoms of pain as well as preventing any complications. Regular visits to hematologist are required to check the blood count and monitor one’s overall health.

Existing Treatments
1. Blood and Marrow Stem Cell Transplant: This treatment may cure a small percentage of people. For this procedure to work, the stem cells used for the transplant should closely match those of the affected person and should be from a family member who doesn’t have sickle cell anemia, thus limiting the patients who may find a donor.
Researchers are also looking for sources of bone marrow stem cells like blood from baby’s umbilical cord etc. Though these studies offer promising steps towards treatment of this disease, they may pose the risks in finding a donor as well as body rejecting the transplant, which may result in life threatening complications.
2. Medications:
- Antibiotics: Children suffering from sickle cell anemia may be required to take antibiotics like penicillins until the age of five years to prevent infections like pneumonia.
- Hydroxyurea: This drug stimulates the production of fetal hemoglobin (a type of hemoglobin that prevents the formation of sickle cells and is found in newborns). But there is a concern that if this drug is taken for a long term, it may cause tumors or leukemia.
- Pain Killers: They are usually prescribed during the periods of crises.
3. Vaccinations:
Though vaccinations in childhood are important for preventing infections in all children, they are even more important for children with sickle cell anemia since even the smallest infection can prove to be severe in them.
4. Blood transfusions:
In blood transfusions, red blood cells from donated blood are given to the affected person intravenously. This increases the normal blood cell count in the body, relieves anemia and decreases the risk of stroke. But this process may carry some risk of increasing the iron levels in the body, which may cause a risk of heart, liver or other organ damage.
Experimental Treatments
1. Gene Therapy:
Gene therapy is being investigated as a possible treatment for sickle cell anemia by exploring the option of inserting a normal gene into the bone marrow which may cause the body to make normal hemoglobin. Researchers are also studying the possibility of "turning off" the sickle hemoglobin gene or the defective gene and reactivating another gene that makes the red blood cells behave normally.
2. New Drugs:
Several medicines and drugs are being studied for the treatment of this disease. These include:
- Decitabine: This drug helps the body to make fetal hemoglobin which will further prevent red blood cells from sickling and improves anemia. This medicine can be used instead of hydroxyurea or may be added to hydroxyurea.
- Adenosine A2a receptor agonists: These medicines reduce complications related to pain in patients already suffering from sickle cell anemia.
- 5-HMF: It is a natural compound that gets attached to the red blood cells and increases their oxygen absorption, thus preventing them from sickling.
- Statins: These medications improve the blood flow through the blood vessels.
See Also: White Blood Cell Count High: Scared? Here's What You Need To Know
3. Nitric oxide: This is a gaseous compound that works as a signal messenger in the body. Nitric oxide reduces the stickiness of the red blood cells and keeps the blood vessels open. People suffering from sickle cell anemia usually have low levels of nitric oxide in their blood. Studies on nitric oxide have shown a mixed result so far but a treatment, if developed, may prevent sickle cells from clumping together.
Sickle cell anemia is an inherited genetic condition and a life-long disease. There is a lot of on-going research on the treatment methods for this disease with the hope that these studies will provide a way to predict the severity of the condition as well as provide a better treatment for the affected patients.
- Gladwin MT, Sachdev V. Cardiovascular abnormalities in sickle cell disease. J Am Coll Cardiol. 2012
- 59(13):1123-33
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010
- 376(9757):2018-2031
- Geller AK, O'Connor MK. The sickle cell crisis: a dilemma in pain relief. Mayo Clin Proc. 2008
- 83:320-323
- Brawley OW, Cornelius LJ, Edwards LR, Gamble VN, Green BL, Inturrisi C, et al. National Institutes of Health Consensus Development Conference statement: hydroxyurea treatment for sickle cell disease. Ann Intern Med. 2008
- 148(12):932-8
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008
- 359(21):2254-65
- Gladwin MT, Kato GJ, Weiner D, Onyekwere OC, Dampier C, Hsu L, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA. 2011
- 305(9):893-902.Photo courtesy of euthman via Flickr: www.flickr.com/photos/euthman/5610746554
- Photo courtesy of Nicolas Alejandro Street Photography via Flickr: www.flickr.com/photos/nalejandro/13200868333
Your thoughts on this